中国科大等在乙肝病毒导致肝癌的免疫逃逸机制研究中取得进展
近日,中国科学技术大学免疫学研究所教授田志刚课题组与山东大学药学院教授张彩课题组合作,揭示了乙肝病毒感染导致机体T淋巴细胞免疫耗竭的新机制。
慢性病毒感染和肿瘤微环境可以诱使机体抗病毒或抗肿瘤特异性T细胞衰竭,使其增殖能力和效应功能严重受损,致使机体免疫应答无法抵抗病毒感染或肿瘤发生。T细胞共抑制受/配体被认为是慢性病毒感染和肿瘤中T细胞耗竭的关键调节因素,据此原理诞生的卡控点免疫疗法(Checkpoint Immunotherapy),被列入2013年度“世界十大科技进展”榜首,成为美国2016年启动的“癌症登月计划(Cancer MoonShot)”的核心手段。然而,慢性病毒感染、肿瘤发生和介导T细胞耗竭的共抑制受/配体三者之间相互影响的分子机制尚不清楚。
在该研究中,科研人员通过HBV+肝癌患者队列进行了肝癌组织的分子病理学研究,发现肝癌细胞的锌指转录因子SALL4(一种肝脏胚胎蛋白)和T细胞共抑制配体PD-L1的表达水平均与miR-200c水平呈明显的负相关。通过大样本肝癌病人生存期回顾分析,发现低表达SALL4或PD-L1及高表达miR-200c的患者具有更长的生存期。以HBV+肝癌细胞为模型探讨其分子机制,发现miR-200c可直接靶向PD-L1的3'-UTR以控制其表达,过表达miR-200c能直接抑制HBV诱导的肝细胞PD-L1表达,证实miR-200c是PD-L1表达的阻遏因子。进一步研究发现,HBV感染可以再激活正常成年肝脏不表达的癌胚蛋白SALL4,而SALL4可特异性负向调控miR-200c的转录,使miR-200c失去对PD-L1表达的阻遏作用,导致PD-L1高表达。应用HBV携带小鼠模型进一步确证,HBV可以导致小鼠肝细胞PD-L1高表达,并伴随抗HBV特异性CD8+ T细胞的耗竭;抑制SALL4表达或增强miR-200c表达或用抗体阻断PD-L1,均能明显削弱PD-L1介导的T细胞耗竭。
该研究首次揭示了在HBV感染和肝癌发展期间存在调节PD-L1表达的HBV-pSTAT3-SALL4-miR-200c-PDL1分子轴,基于该分子轴的干预策略对逆转病毒或肿瘤诱导的免疫细胞功能衰竭具有重要意义,为Checkpoint免疫治疗提供了新思路。同时,该研究还揭示了miR-200c和SALL4可能对HBV+肝癌患者的预后具有预测价值,具有重要的分子病理学意义。
相关研究成果发表在《自然-通讯》上,中国科大副教授孙成、山东大学博士兰培祥和副教授韩秋菊为论文共同第一作者。该研究得到了国家自然科学基金的资助和华中科技大学教授张智红、安徽省立医院黄强等的研究支持。
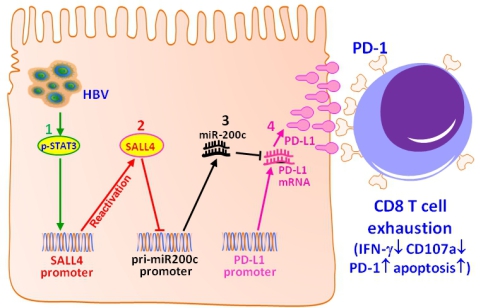
HBV感染通过HBV-pSTAT3-SALL4-miR-200c-PD-L1诱导T细胞耗竭